Blast 10 Ways
Adventures in bioinformatics tool deployment

Waiting for Feedback
This project started when a student asked “Do I really have to SSH into the main server each time I want to run an updated version of my script?”. She asked because her task was to modify an existing analysis script for a bioinformatics research project.
I set out to work to find out how my student could get faster feedback as she made changes to the Python script and this is where that journey led…
1. Run it Locally
I started work on this challenge with a pairing partner. He and I first examined our student’s script and found that the script was using a standard bioinformatics tool —NCBI blast(Basic Local Alignment Search Tool). So, we wondered, should our student just install the binary for this tool on her laptop?
Blast finds regions of similarity between biological sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance.
Hmm…because we didn’t know how blast was supposed to work, we thought we’d like to have a quick ‘hello world’-type example as we tested various types of deployments. Also, we wanted to run a bash script which downloads the blast binary and then runs an example analysis from within a Jupyter notebook. We preferred this to installing blast in the container, as it seemed simpler. We used a notebook so we could add notes (in markdown) and visualizations.

We googled and found examples for using blast and Jupyter in docker containers. We built our combined service DOCKERFILE and an iPython example notebook file, then we spun up a local instance of the container and …success! (images shown left and below).
We published our work on GitHub and as docker container image on DockerHub.

Our student was now happy, but we were still curious about other possible ways to deploy this set of tools…
Next we wanted to explore running blast on the public cloud. We also wanted to use our ‘hello blast’ example when we tried out deployment / pipeline APIs and tools for building and deploying more complex bioinformatics research data pipelines.
2.-10. Running Elsewhere…cloud and…
As a simple place to start we wanted to run our example container image on a GCP Virtual Machine.
2. First we ran our blast container image on a GCE VM instance optimized for running containers. To do so, we simply checked the ’deploy a container image to this VM instance’ option on GCE & filled in the ‘Container image’ field as shown below — DockerHub, ‘registry.hub.docker.com/lynnlangit/blastn-jupyter-docker’). Docker tools preinstalled — Simple and good.

3. Next we wanted to use the newly-released GCP AI Hub Notebooks. We started a Python-based AI Hub Notebooks VM instance, clicked the ‘OPEN JUPYTERLAB’, link (shown below). Then we uploaded the example blast notebook which downloads and runs blast. This was quick and simple — no container image needed, just our example notebook.


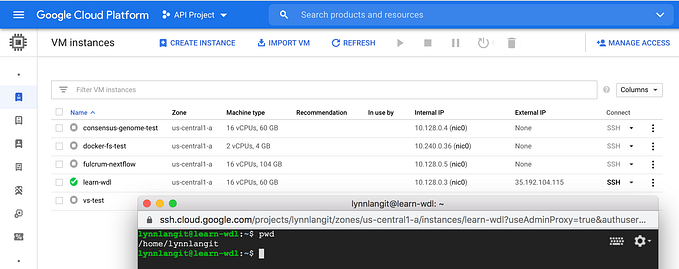
4. Next we wanted to run our container on GKE in a Kubernetes cluster. We selected ‘deploy a container’ and quickly created our deployment.

5. Next we wanted to run consider running blast on GCE w/Google Pipelines API, cromwell and WDL. We looked at the Pipelines API sample (using best practice for The Broad’s GATK tools) and decided that we didn’t need the distributed compute for our workload type.
6. We also wanted to consider running blast on GCE using the job submission dsub command-line tool. An alternative to the gcloud tool, dsub adds user interfaces and workflow definition features that supplement the GCP Pipelines API. We reviewed the sample (which uses a container with samtools) and decided this would be overkill for our simple process. Shown below is a sample job script.

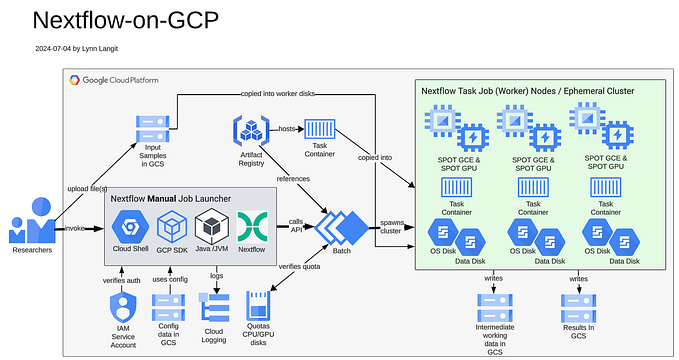
7. Next we wanted to run blast w/ Nextflow — we started by running their blast sample locally. This was simple to do, we just followed Nextflow’s Quick start instructions (shown below). Their example includes a reference to docker container image in their GitHub repo, so to use our container, we could just switch out the value in the nextflow.config file. Nextflow pipelines can be run on GCP as well.

8. Next we wanted to run blast on Galaxy Project by running an example workflow or by getting getting blast from the Galaxy Tool shed. We saw that Galaxy includes a number of tools which implement various version of blast, so we concluded that if we wanted to run blast on Galaxy we’d just use their tools (shown below) and their public interface (the UseGalaxy website) shown below.

9. Next we wanted to run blast on Terra.bio (was Firecloud, runs on GCP). We first ran on Terra.bio as a Jupyter notebook which installs blast. We created a workspace in Terra, then uploaded the example notebook in the Notebooks section. We started a notebook runtime instance in Terra and ran the notebook — simple! Note that you can configure the size of the notebook runtime (amount of CPU, etc..) by clicking on the gear in the upper right before you click on the start (blue triangle).


10. We next wanted to run our blast container image on Terra.bio as a job. We had to first create two WDL files (one for blastn as a tool and one for blastn as a single-tool workflow) to register our blast container image as a workflow in Dockstore.
We then selected the ‘public view’ of the workflow in Dockstore and then clicked on the ‘launch with Terra” button in Dockstore to add our blast workflow to the Terra.bio site as a executable job/workflow. See screenshots below for job flow. Terra workflows use the cromwell engine and also the Google Genomics / Pipelines API.



What We Learned
What started as a simple experiment (i.e. containerize and run a bioinformatics tool locally) turned into a long, but fun set of deployment projects!
We learned there are so many tools and options in the bioinformatic space — -this makes selecting a best-fit deployment method really challenging for analysis. We really like having our ‘blast’ (hello-world) example, as it allows us to try out different techniques quickly. Also, we find ourselves using Jupyter notebooks for quick prototyping and testing frequently.
In order to map the landscape of bioinformatics tools, APIs and cloud vendors, I am working on a MindMap (screen below for GCP) and also a GitHub repository with one-page explanations and short screencasts of key GCP services to help to mitigate the challenges of navigating this complex landscape.

We would be interested in hearing from others in the bioinformatics community. Which deployment methods work best for you and your research teams?
#HappyDeploying
Resources
- DOCKERFILE with notebook that downloads blast on GitHub
- Jupyter container downloads blast on Dockerhub
- DOCKERFILE includes blast on GitHub
- Blast container image on DockerHub
- Blast WDL tool and workflow on Dockstore
- Nextflow blast pipeline on Nextflow
- GCP-for-Bioinformatics one page explanations and screencasts on GitHub